Last updated: April 29, 2026
Medically reviewed by: NKF Patient Education Team
Fabry disease is a rare inherited condition that can affect many parts of the body, including the kidneys. Learn more about symptoms, causes, diagnosis and treatment.
Signs and Symptoms
Symptoms may vary depending on different things, such as age or severity of the disease. Not everyone with Fabry disease will have all the symptoms and complications. For example men tend to have more severe Fabry disease symptoms than women. In addition, symptoms can change if the disease advances. Signs and symptoms for Fabry disease can include:
- Pain: Severe burning pain in hands and feet (acroparesthesias).
- Less sweating: Can include sweating less (hypohidrosis) or not at all (anhidrosis).
- Heat or cold intolerance: Heat or cold can trigger pain in hands and feet. Warmer weather and more physical activity can cause overheating, frequent fevers and pain sensitivity.
- Skin rash: Red or purple rashes (angiokeratomas), often around the belly button, buttocks, groin or upper thighs.
- Corneal pattern (whorls): A starburst pattern found on the cornea (front surface of the eye). The pattern can be found by an eye exam by an ophthalmologist (eye doctor). It usually does not affect vision.
- Gastrointestinal (stomach and intestine) problems: Can include diarrhea, nausea, constipation and abdominal pain.
- Hearing problems: Can include hearing loss or ringing in ears (tinnitus).
Symptoms from kidney disease caused by Fabry disease can include:
- High protein in urine (proteinuria): Happens when too much protein leaks into the urine. This may cause the urine to look foamy and/or cloudy.
- Swelling (edema): Usually happens around the hands, feet, and ankles. There can also be puffiness around the eyes.
Causes
Fabry disease is an inherited disorder, which means it is passed from parents to children. It is caused by variants (changes) in the GLA gene, which is located on the X chromosome. This means that Fabry disease has an X-linked inheritance pattern. As a result, fathers can pass along an X chromosome with the GLA gene variant to all of their daughters, but none of their sons. Mothers have a 50% chance of passing along an X-chromosome with the GLA gene variant to their daughters or sons.
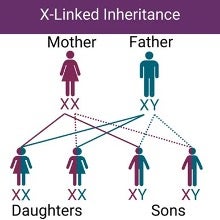
Because of the way the disease is inherited, men often have more severe symptoms. Women can also be affected, but symptoms can vary from mild to severe. The differences in severity among women is from a process called X-inactivation (lyonization). This is a regular biochemical process where one of the X chromosomes is turned off in cells. During this process, a GLA gene variant that may cause Fabry disease can be turned on or off.
The GLA gene codes for an enzyme (type of protein) called alpha-galactosidase A (alpha-GAL). This enzyme breaks down certain fats so they can be removed from cells and passed out of the body, or be recycled for other uses. A GLA gene variant can lead to alpha-GAL enzymes that are either missing or present in low amounts. It can also lead to enzymes that are not able to work the way they should. Because of this lower alpha-GAL enzyme activity, fat builds up in cells of the blood vessels and tissues of the skin, brain, heart and kidneys. Over time, this buildup can lead to problems with many parts of the body, including the brain and nervous system, skin, heart, and kidneys.
Because Fabry disease affects multiple parts of the body, it is known as a multi-systemic disease.
Types
Types of Fabry disease include the following:
- Early onset (Classic) type: Symptoms appear during childhood or teenage years. Tends to advance faster and have more severe multi-system symptoms.
- Late onset (non-classical or atypical) type: Symptoms later in adult life (30’s or older). Tends to advance slower. Fewer parts of the body may be impacted, but the heart and kidneys can still be affected.
Fabry Disease
Sign up for a deep dive into Fabry Disease
Learn about Fabry disease, receive additional resources, and learn so much more.
Complications
Complications of Fabry disease can include:
- High blood pressure (hypertension)
- High cholesterol
- Nervous system problems
- Hearing problems
- Stomach problems
- Heart attack or stroke
- Kidney failure
Living with a chronic illness like Fabry disease can be physically and emotionally challenging. Fabry disease can cause multiple emotional challenges. Many people with Fabry disease may have feelings of fear, distress, anxiety, or depression, related to their illness.
Diagnosis
Diagnosis may be difficult because Fabry disease is a rare disease. If you have one or more of the symptoms listed above, you should speak to your healthcare team. After an examination, your healthcare team will decide whether certain tests are needed. Fabry disease is diagnosed through an enzyme assay (for men) or genetic testing (for men and women).
Tests
Tests for Fabry disease can include:
- Enzyme assay: Measures the amount of alpha-GAL enzyme activity in your blood. Lower enzyme activity (result of 1% or less) usually means Fabry disease. This test is more reliable for men. However, the test is not as reliable or accurate for women.
- Genetic testing: The test samples of blood or saliva, which are sent to a lab to look for GAL gene variants. The genetic test may be a more accurate test in women who may have a milder form of the disease. A physician or genetic counselor can order genetic testing. A genetic counselor is a healthcare professional with special training in genetics and genetic diseases. They can help answer questions about the test and its results.
A skin biopsy or kidney biopsy can also help establish the diagnosis. Biopsies can also be used to monitor how the disease is advancing, or see how well a certain treatment is working.
If you or a family member has been diagnosed, others in your immediate and extended family may also have Fabry disease. Talk to your doctor or a genetic counselor about family testing.
Treatment
There is no cure for Fabry Disease. So, treatment is focused on managing symptoms, and managing your heart and kidney health. Medications that directly target the Fabry disease process are available, with more being studied in clinical trials. Fabry disease has a lifelong impact, so it is important to keep up with medical appointments, take your medicines, and stay with the treatment plan as recommended by your healthcare team.
Medications
Medications that directly target deficient alpha-GAL activity in Fabry disease are available. FDA-approved treatments for Fabry disease include enzyme replacement therapy and chaperone therapy.
Enzyme Replacement Therapy (ERT) replaces the missing enzyme, alpha-GAL, in Fabry disease.
- Agalsidase beta (Fabrazyme) is an intravenous (IV) ERT approved for people with Fabry disease 2 years of age and older. It is given as an IV infusion every two weeks. It contains the alpha-GAL enzyme that is deficient in Fabry disease.
- Pegunigalsidase alfa (Elfabrio) is an IV ERT approved for adults with Fabry disease. It is given as an IV infusion every two weeks. It has the alpha-GAL enzyme, modified with polyethylene glycol (PEG). This may make the drug stay in the body longer, which may allow for shorter infusions or less frequent infusions. The exact infusion schedule and dosing will be carefully decided by your healthcare team.
Chaperone therapy uses small molecules to correct or stabilize existing alpha-GAL.
- Migalastat (Galafold) is approved for adults with Fabry disease. It is an oral medication (taken by mouth). This medication is only for people with an amenable GAL gene variant. This means the medication is for some, but not all, people with certain GAL gene variants linked with Fabry disease. A genetic test is used to see if a person with Fabry disease has an amenable gene variant for this treatment. Migalastat acts as a “chaperone” to help the alpha-GAL enzyme work the way it should.
Other treatments may also be used to manage certain problems or symptoms.
- Heart disease and stroke: As in people without Fabry disease, treatment depends on the specific heart problem and may include medications, surgery or pacemakers. To help prevent strokes, medications to keep blood from clotting may be used.
- Kidney disease: If mild, kidney problems may be addressed with low-salt diets and certain medications. For example, special medications called ACE inhibitors or ARBs can be used if blood pressure is high, or if there is protein in the urine (proteinuria). If kidney failure happens, then a kidney transplant or dialysis is needed.
- Pain: People with Fabry disease may need to limit strenuous activities, drink more water and other fluids and take frequent naps to lessen fatigue. Pain-relieving medications may also help. Some pain medications may hurt your kidneys, so speak with your healthcare professional to discuss pain treatments that may be right for you.
- Stomach issues: Your healthcare team may recommend nutritional changes or medications. Speak with your health professional if you experience nausea, diarrhea, pain, cramping, or other stomach issues.
Nutrition
Practicing a healthy lifestyle should also be part of the treatment plan. You may need to limit your sodium, or salt intake, especially if you have high blood pressure. You may also be asked to lower the amount of fat you eat, or avoid certain foods that can create stomach problems. You may be asked to change your diet in other ways. A dietitian can help with a meal plan that’s right for you.
Exercise
Exercise and physical activity are important parts of your health. It can also help with heart and kidney health. However, exercise can be a challenge for people with Fabry disease, especially if they have pain, temperature sensitivity, or reduced sweating. This can make exercise very difficult.
Don't confuse physical activity with vigorous exercise. Any type of body movement helps including walking or doing chores. The key is to find something that you enjoy and works best for you. Lower impact exercises or training may be advised by your healthcare team. Check with your healthcare team before starting any exercise and ask which exercises are best for you.
Preparing for your appointment
Questions to ask
- Do I need a referral for a genetic counselor?
- Is genetic testing covered by my health plan?
- Should I or my family be tested for Fabry disease?
- What is my risk of developing kidney failure because of my Fabry disease?
- What is my risk for heart disease because of my Fabry disease?
- What lifestyle changes do you suggest I make to help prevent complications?
- Are there any changes I need to make with my diet? Can a dietitian help me with my diet?
- Do I have to make any changes with my medications?
- What symptoms should I watch for that might indicate my condition is worsening?