Evaluación y tratamiento de la cistinosis nefropática, incluyendo el diagnóstico, el tratamiento con cisteamina y las estrategias de atención a lo largo de toda la vida.
Fisiopatología
La cistinosis nefropática es un trastorno metabólico autosómico recesivo. Se trata de una enfermedad rara que afecta al paciente durante toda su vida. La incidencia anual de la cistinosis nefropática es de aproximadamente 1 por cada 150,000 – 200,000 nacidos vivos, y su prevalencia es de aproximadamente 1.6 por cada millón de habitantes.1 Sin embargo, se ha observado que la incidencia es mayor en determinadas poblaciones a nivel mundial.2, 3
La cistina se deriva de la degradación enzimática normal de las proteínas en los lisosomas de la célula. La cistina es transportada a través de la membrana lisosómica hacia el citosol por la cistinosina, una proteína transportadora específica de la membrana lisosómica.3,4
La cistinosis se caracteriza por una acumulación del aminoácido cistina en todo el organismo, como consecuencia de un transporte deficiente de este fuera de los lisosomas de las células.4 La cistinosis nefropática está relacionada con más de 100 mutaciones conocidas en el gen CTNS, que codifica la cistinosina, y da lugar a la acumulación de cistina intralisosómica en numerosos órganos y tejidos (Figura 1).1,4
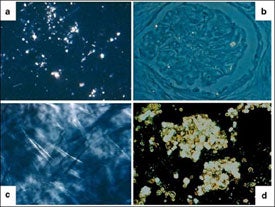
con cistinosis nefropática (a) hígado; (b) riñón; (c) córnea; (d) médula ósea
La mutación más frecuente conocida asociada a la cistinosis entre los europeos del norte es una deleción de 57 kb que afecta a los primeros 10 exones del gen CTNS y a otros dos genes: CARKL y TRPV1.6,7,8 El gen CARKL (similar a la quinasa de carbohidratos) codifica la enzima sedoheptulocinasa, y el gen TRPV1 (miembro 1 de la subfamilia V de los canales catiónicos de potencial receptor transitorio) codifica el receptor de potencial transitorio vaniloide.
Presentación clínica
La cistinosis presenta tres formas clínicas, según la edad en la que aparecen los primeros síntomas: nefropática infantil clásica/de inicio precoz; nefropática intermedia/de inicio tardío; y no nefropática en adultos/ocular.
La cistinosis nefropática infantil es la forma más frecuente de la enfermedad y representa aproximadamente el 95% de los casos. Suele presentarse en la primera infancia junto con el síndrome de Fanconi, que consiste en múltiples defectos en la reabsorción tubular proximal renal. Si no se trata, el síndrome de Fanconi conduce a falla renal (enfermedad renal terminal oERT) en los últimos años de la infancia.1,9, 10
Los niños que nacen con cistinosis nefropática suelen ser asintomáticos y presentar un peso y una estatura normales. Los primeros signos clínicos apreciables de la cistinosis infantil suelen aparecer entre los 6 – 12 meses de edad. Los síntomas pueden incluir retraso en el crecimiento, intolerancia alimentaria y episodios de hipovolemia (deshidratación). Estos síntomas se deben, en parte, al síndrome de Fanconi renal, que se caracteriza por una pérdida urinaria excesiva de sodio, potasio, fosfato, calcio, magnesio y agua, junto con una disminución de la activación del calcitriol, un metabolito de la vitamina D producido por el riñón. Estas anomalías metabólicas pueden provocar hipopotasemia, hiponatremia, acidosis metabólica, escaso aumento de peso, crecimiento lineal deficiente, raquitismo óseo y/o debilidad muscular generalizada. Otros órganos y tejidos también pueden verse afectados por la cistinosis, lo que da lugar a complicaciones retinianas, endocrinas y neuromusculares a medida que el paciente envejece. Los jóvenes con cistinosis también pueden presentar problemas neurocognitivos, como una integración visomotora deficiente, dificultades para mantener la atención, problemas de memoria visual y de la función ejecutiva.
Por lo general, en la forma infantil de la cistinosis nefropática, la función de filtración renal se deteriora lentamente hasta los 5 años de edad, pero la pérdida de función puede acelerarse a partir de entonces en un paciente no tratado, lo que conduce a falla renal alrededor de los 10 – 12 años de edad (Figura 2).11,12 Antes de la aparición de los tratamientos de depleción de cistina y del trasplante renal, la esperanza de vida de un niño con cistinosis nefropática era de unos 10 años. Sin embargo, gracias a una atención médica moderna y óptima, los pacientes viven actualmente hasta mediados de los cuarenta o los cincuenta con una calidad de vida satisfactoria, y los resultados a largo plazo están mejorando.1
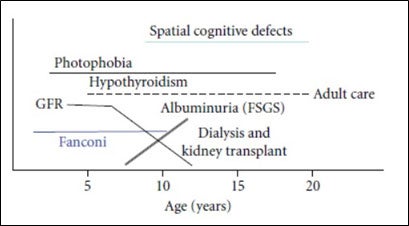
Aproximadamente el 5% de los casos de cistinosis corresponden a la forma nefropática juvenil (de aparición tardía). Estos pacientes no presentan síntomas clínicos apreciables hasta finales de la infancia o la adolescencia. Las manifestaciones clínicas pueden incluir un síndrome de Fanconi leve o ausente y proteinuria asintomática. Pueden presentar síndrome nefrótico, que puede confundirse con otras causas.14 Por lo general, estos pacientes presentan una progresión más lenta hacia la falla renal.
La forma ocular de la cistinosis en adultos, no nefropática, suele afectar únicamente a los ojos. Se caracteriza por fotofobia debida a la acumulación de cistina en la córnea y rara vez se manifiesta antes de la edad adulta.2
Diagnóstico
La detección de niveles elevados de cistina en los glóbulos blancos (GB) se considera la principal prueba diagnóstica de la cistinosis. También puede utilizarse para evaluar la eficacia del tratamiento de eliminación de cistina. A lo largo del tiempo se han desarrollado múltiples métodos para medir la cistina en los glóbulos blancos, entre los que se incluyen los ensayos de proteínas de unión a la cistina, la cromatografía líquida de alta resolución (HPLC) o la cromatografía líquida acoplada a espectrometría de masas en tándem (LC-MS/MS).1,15,16,17 La espectrometría de masas en tándem se considera actualmente el método de elección.1, 10
Los pacientes recién diagnosticados con cistinosis nefropática presentarán casi siempre niveles de cistina en los leucocitos comprendidos entre 3 – 20 nmol de semicistina/mg de proteína.1, 10 Se ha sugerido que estos niveles elevados de cistina en los leucocitos, a cualquier edad, constituyen un diagnóstico definitivo. Los individuos de control y los portadores heterocigotos presentan niveles que oscilan entre <0.2 – <1.0 nmol de semicistina/mg de proteína, respectivamente.1, 10
El análisis molecular del gen CTNS se considera importante en el asesoramiento genético y, por lo general, se recomienda para confirmar un diagnóstico inicial.1 No se incluye en la evaluación neonatal habitual. Sin embargo, puede considerarse la realización de la prueba del gen CTNS en un recién nacido o feto con antecedentes familiares muy marcados. La prueba del gen CTNS puede revelar aproximadamente el 95% de las mutaciones causantes de la enfermedad.10
La detección de cristales de cistina en la córnea mediante examen con lámpara de hendidura es otra opción posible para confirmar el diagnóstico. Sin embargo, los cristales de cistina en la córnea no aparecen con este método hasta el segundo año de vida. Este examen debe ser realizado por un oftalmólogo familiarizado con la enfermedad para identificar y clasificar adecuadamente la carga de cristales en la córnea.10
El análisis de orina en pacientes con cistinosis nefropática puede mostrar una baja gravedad específica, glucosuria manifiesta (con glucosa sérica normal) y albuminuria de leve a moderada.2 La creatinina sérica suele ser normal en niños pequeños, a menos que estén deshidratados. Dado que muchos niños con cistinosis presentan atrofia muscular y retraso en el crecimiento, la creatinina sérica por sí sola (que es un producto de la degradación muscular) puede ser un mal marcador de la función renal.
La cistinosis debe tenerse en cuenta en el diagnóstico diferencial de todo paciente joven que presente falla renal de origen desconocido, especialmente si hay antecedentes de poliuria y retraso en el crecimiento.10
Gestión
En general, el tratamiento de la cistinosis nefropática consiste en la administración de cisteamina por vía oral y otros tratamientos para controlar el síndrome de Fanconi, incluido el control de los líquidos y los electrolitos. Cualquier plan de tratamiento debe someterse a evaluaciones periódicas y revisarse para mejorar la atención y abordar los problemas que surjan a lo largo de la vida del paciente, a medida que sus necesidades cambian con el tiempo.1, 10,18
Los lactantes diagnosticados con cistinosis nefropática suelen necesitar visitas más frecuentes al nefrólogo durante la primera infancia y la niñez que durante la adolescencia. Por lo general, es necesario controlar los niveles de cistina en los glóbulos blancos y otros parámetros sanguíneos cada 3 – 4 meses. Otros componentes importantes del tratamiento de la cistinosis nefropática en lactantes y niños incluyen ajustes nutricionales adecuados, así como el control de líquidos y electrolitos (dependiendo de la presencia y la gravedad del síndrome de Fanconi).
A medida que un niño con cistinosis nefropática entra en la adolescencia, comienzan a surgir otras cuestiones y retos especiales, entre los que se incluyen el cumplimiento de los tratamientos prescritos, el aumento de la probabilidad de desarrollar algunas complicaciones extrarrenales (p. ej., hipotiroidismo, diabetes mellitus de tipo I que requiere insulina, complicaciones neuromusculares), la progresión hacia la falla renal con la necesidad de terapia de sustitución renal, y la transición a la edad adulta y a la atención médica para adultos.19, 20
A medida que los pacientes con cistinosis viven más tiempo en la edad adulta, se han observado más complicaciones a largo plazo, ya que no tuvieron tiempo suficiente para desarrollarse en la etapa pediátrica debido a la ausencia de estas terapias. Además, sigue existiendo la posibilidad de que se desarrollen manifestaciones aún no descritas en pacientes que han recibido un tratamiento precoz y adecuado.1, 10
Se recomienda un equipo multidisciplinar para los pacientes con cistinosis, que incluya, cuando proceda, especialistas en nefrología/nefrología de trasplantes, neurología, oftalmología, neumología, endocrinología, trabajadores sociales, psicólogos, otorrinolaringólogos especializados en disfunciones de la deglución, logopedas y fisioterapeutas.
Terapia de reducción de la cistina
El pilar fundamental del tratamiento de la cistinosis nefropática es la terapia de reducción de la cistina con compuestos de bitartrato de cisteamina. Dicha terapia reduce la cistina lisosómica mediante la formación de un complejo disulfuro mixto de cisteamina y cistina que sale del lisosoma a través de un transportador PQLC2 intacto (Figura 3).10
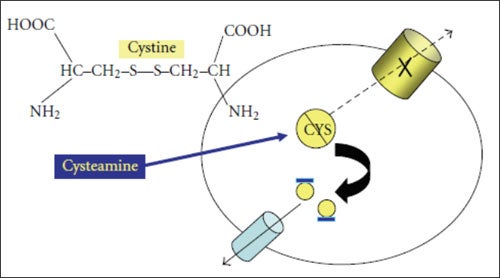
La preparación original de bitartato de cisteamina se denomina ahora de liberación inmediata. Con esta preparación, la dosis solía ser de 60 – 90 mg/kg al día o de 1.35 – 1.90 g/m² de superficie corporal al día, administrada cada seis horas. Esta formulación estaba disponible en EE. UU. y en gran parte del continente europeo desde la década de 1990.
En 2013, la FDA aprobó una formulación de bitartrato de cisteamina de liberación retardada, que se administra cada doce horas, para el tratamiento de la cistinosis nefropática en adultos y pacientes pediátricos. En 2015, la aprobación de la FDA se amplió para niños de ≥6 años de edad a ≥2 años de edad. Esto reduce la frecuencia de dosificación de cada seis horas a cada 12 horas al día.18 Un ensayo clínico prospectivo y controlado demostró una eficacia farmacodinámica continuada tras 24 meses de tratamiento.21
El tratamiento tópico con colirio de clorhidrato de cisteamina se administra para tratar los cristales de cistina en la córnea.18 En 2012, la FDA aprobó una solución oftálmica comercial de cisteamina al 0.44% para el tratamiento de los cristales de cistina en la córnea en pacientes con cistinosis.
Terapia de sustitución renal
El síndrome de Fanconi renal puede persistir tras el inicio de la diálisis o tras un trasplante renal en los riñones nativos, pero rara vez requiere nefrectomías de los riñones nativos, ya que las pérdidas excesivas de líquido y electrolitos suelen reducirse con la terapia de sustitución renal.22 La presencia de poliuria y pérdida de sodio limita la necesidad de ultrafiltración en pacientes con cistinosis sometidos a hemodiálisis o diálisis peritoneal que conservan función renal residual.1,23
El trasplante renal es el tratamiento preferido para la falla renal en la cistinosis nefropática. Tanto los riñones de donantes vivos como los de donantes fallecidos son eficaces. En general, el trasplante renal se asocia a mejores resultados en la cistinosis nefropática que en pacientes sometidos a trasplante por otras indicaciones; no obstante, se requiere una vigilancia continua.20,22,24
El trasplante renal no corrige el defecto metabólico en otros tejidos, y la cistina sigue acumulándose y causando daño orgánico. Por lo tanto, el tratamiento con cisteamina continúa de por vida tras el trasplante. El objetivo del tratamiento con cisteamina tras el trasplante es retrasar o revertir el daño en los órganos extrarrenales.20,22
Es fundamental que los profesionales de la salud comprendan que la cistinosis de un paciente no se cura tras el trasplante y que la atención multidisciplinar es esencial para promover la salud a largo plazo.22 La adherencia a los tratamientos prescritos para la cistinosis, las afecciones relacionadas y/o un trasplante renal puede suponer un reto constante para estos pacientes, que deben gestionar un régimen terapéutico complejo a lo largo de toda su vida.
Referencias
- Langman C, Barshop B, Deschênes G, et al. Controversias y agenda de investigación en la cistinosis nefropática: conclusiones de una conferencia sobre controversias organizada por “Enfermedad del Riñón: Mejora de los Resultados Globales” (KDIGO). Kidney Int. 2016;89:1192 – 1203.
- Niaudet P. Cistinosis. Matoo T (ed.). UpToDate, Waltham, MA. Consultado el 6 de julio de 2017.
- Gahl W, Thoene J, Schneider J. Cistinosis. N Engl J Med. 2002;347:111 – 211.
- Gahl W, Thoene J, Schneider J. Capítulo 199. Cistinosis: un trastorno del transporte de la membrana lisosómica. En: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. Las bases metabólicas y moleculares de las enfermedades hereditarias, octava edición. McGraw-Hill Companies; 2001:5085 – 5108.
- Nesterova G, Gahl W. Cistinosis nefropática: complicaciones tardías de una enfermedad multisistémica. Pediatr Nephrol. 2008;23:863 – 878.
- Taranta A, Wilmer M, van den Heuvel L, et al. Análisis de los transcritos del gen CTNS en la cistinosis nefropática. Pediatr Nephrol. 2010;25:1263 – 1267.
- Topaloglu R, Vilboux T, Coskun T, et al. Bases genéticas de la cistinosis en pacientes turcos: experiencia de un único centro. Pediatr Nephrol. 2012;27:115 – 121.
- Wamelink M, Struys E, Jansen E. La deficiencia de sedoheptulocinasa debida a una deleción de 57 kb en pacientes con cistinosis provoca la acumulación urinaria de sedoheptulosa: elucidación del gen CARKL. Hum Mutat. 2008;29:532 – 536.
- Ivanova E, De Leo MG, De Matteis MA, Levtchenko E. Cistinosis: presentación clínica, patogénesis y tratamiento. Pediatr Endocrinol Rev. 2014;12(Suplemento 1):176 – 184.
- Elmonem M, Veys K, Soliman N, van Dyck M, van den Heuvel L, Levtchenko E. Cistinosis: una revisión. Orphanet J Rare Dis. 2016;11:47.
- Greco M., Brugnara M., Zaffanello M. y cols. Resultados a largo plazo de la cistinosis nefropática: experiencia de 20 años en un único centro. Pediatr Nephrol. 2010;25:259 – 2467.
- Lucky A, Howley P, Megyesi K, et al. Estudios endocrinos en la cistinosis: hipotiroidismo primario compensado. J Pediatr. 1977;91:201 – 210.
- Goodyer P. Historia de la cistinosis: lecciones para el manejo clínico. Int J Nephrol. 2011;2011:929456.
- Servais A, Morinière V, Grünfeld J, et al. Cistinosis nefropática de aparición tardía: presentación clínica, evolución y genotipado. Clin J Am Soc Nephrol. 2008;3:27 – 35.
- Oshima R, Willis R, Furlong C, Schneider J. Ensayos de unión de aminoácidos. Utilización de una proteína de unión a la cistina procedente de Escherichia coli para la determinación de la cistina soluble en ácido en pequeñas muestras fisiológicas. J Biol Chem. 1974;249:6033 – 6039.
- de Graaf-Hess A, Trijbels F, Blom H. Nuevo método para la determinación de cistina en leucocitos y fibroblastos. Clin Chem. 1999;45:2224 – 2228.
- Chabli A, Aupetit J, Raehm M, Ricquier D, Chadefaux-Vekemans B. Determinación de la cistina en granulocitos mediante cromatografía líquida acoplada a espectrometría de masas en tándem. Clin Biochem. 2007;40:692 – 698.
- Emma F, Nesterova G, Langman C, et al. Cistinosis nefropática: documento de consenso internacional. Nephrol Dial Transplant. 2014;29(suplemento 4):iv87 – iv94.
- Brodin-Sartorius A, Tete M, Niaudet P, et al. El tratamiento con cisteamina retrasa la progresión de la cistinosis nefropática en adolescentes mayores y adultos. Kidney Int. 2012;81:179 – 189.
- Van Stralen K, Emma F, Jager K, et al. Mejora del pronóstico renal en la cistinosis nefropática. Clin J Am Soc Nephrol. 2011;6:2485 – 2491.
- Langman C, Rioux P, Greenbaum L, et al. El tratamiento prolongado de pacientes con cistinosis y ERC con RP103 demuestra su eficacia y seguridad. Pediatr Nephrol. 2013;28:1363.
- Wilmer M, Schoeber J, van den Heuvel L, Levtchenko E. Cistinosis: herramientas prácticas para el diagnóstico y el tratamiento. Pediatr Nephrol. 2011; 26:205 – 215.
- Langlois V, Geary D, Murray L, et al. La poliuria y la proteinuria en la cistinosis no influyen en el trasplante renal: un informe del Estudio Cooperativo Norteamericano de Trasplante Renal Pediátrico. Pediatr Nephrol. 2000;15:7 – 10.
- Ehrich JH, Bordehl J, Byrd D, et al. Trasplante renal en 22 niños con cistinosis nefropática. Pediatr Nephrol. 1991;5:708 – 714.